AlphaGenome Coverage Explorer: Check Training Data Coverage Before You Run the Model
A practical tool to check whether your tissue or cell type is covered in AlphaGenome training tracks before you run variant effect analyses.
Before you run AlphaGenome on your biological question, there is one thing worth checking first: whether your tissue or cell type was actually in the model's training data.
AlphaGenome predicts regulatory consequences of DNA variants, gene expression, chromatin accessibility, splicing, 3D genome contacts, across thousands of cell types and tissues. But those predictions are learned from experimental data. If your biosample has sparse coverage or no coverage at all, the model is extrapolating from whatever related tissue it was trained on. That may be fine for your use case. Or it may not. Either way, you should know before you commit to an analysis.
What the tool does
The AlphaGenome Coverage Explorer lets you search 714 human and 179 mouse biosamples by name or ontology term and see exactly what training data exists for each one, track counts broken down by modality, which of the 11 output types are covered and the ontology identifier to use in your API calls.
Search "liver" and you get every liver-related biosample in the training set, primary tissue samples alongside heavily-profiled cell lines like HepG2 (565 tracks across 9 modalities). Search "pancreas" and you immediately see that coverage is sparse: the best-covered pancreatic biosample has 20 tracks across 8 modalities. That context changes how you interpret results.
Toggle between human and mouse datasets. The model was trained on 5,563 human tracks and 1,038 mouse tracks, the explorer covers both, with full per-modality breakdowns for each species.
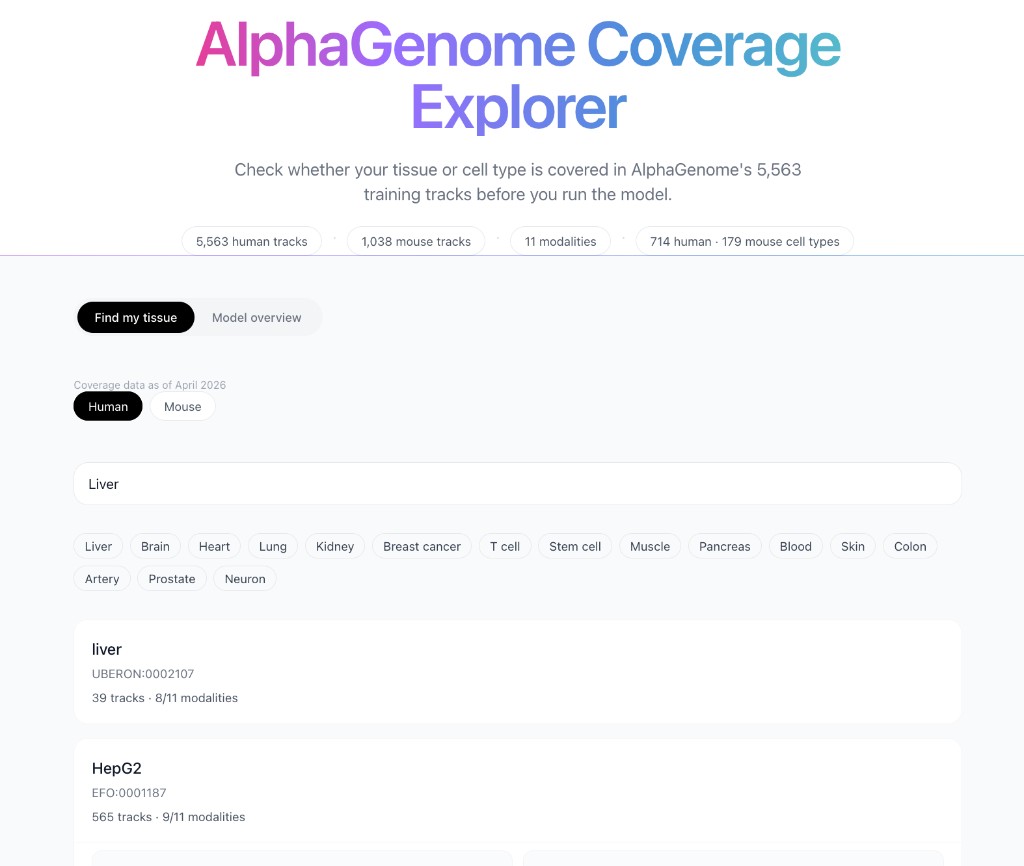
AlphaGenome Coverage Explorer with biosample-level modality coverage and track counts.
How it was built
The data is pulled directly from the AlphaGenome Python SDK using output_metadata(), the same endpoint you would call before running the model in your own pipeline. A Python script fetches the metadata, extracts per-biosample track counts across all 11 modalities and saves the result as a static JSON file. The frontend is a Next.js app that reads from that JSON.
The fetch script and the raw JSON output are both in the GitHub repo. If you want to verify the numbers, regenerate the data with a newer API version or adapt the script for your own tooling, everything is there.
Coverage data was last generated in March 2026 and reflects the AlphaGenome API at that time. The app displays the generation date in the header so you always know what version of the metadata you're looking at.
Where to go from here
If you want to understand how AlphaGenome actually works before running it, the blog series covers the full picture:
- Part 1: The Biology and the Problem: cis-regulatory elements, the long-range versus resolution tradeoff, CNNs and transformers as genomic tools
- Part 2: The Architecture, the Model and What It Can Do: the U-Net, the two-stage distillation training that makes variant scoring reliable and the TAL1 leukemia case study
Part 3, which walks through a saturation mutagenesis analysis on a pharma-relevant enhancer using the API, is coming next.
The explorer is free to use. AlphaGenome is developed by Google DeepMind and is available for non-commercial research via their public API.