Understanding AlphaGenome, Part 1: The Biology and the Problem
From a missing pancreas to cis-regulatory logic: why most disease variants sit outside genes and the range-versus-resolution problem AlphaGenome was built to solve.
This is the first post in a three-part series on AlphaGenome. Part 1 lays the conceptual groundwork, the biology you need, the computational ideas behind it and the core tension the model was built to resolve. Part 2 walks through the AlphaGenome architecture itself. Part 3 shows you how to run it.
A baby was born with no functioning pancreas.
Then another. Then another.
Ten families, across multiple countries, presenting with the same devastating diagnosis: neonatal diabetes requiring insulin from birth, no exocrine pancreatic function, no explanation.[1] Researchers sequenced every gene. Every protein looked normal.
The answer wasn't in a gene at all.
A team led by researchers at the University of Exeter used epigenomic data from embryonic pancreatic progenitor cells to guide their search through whole genome sequences. What they found was a previously uncharacterized 400-base-pair stretch of DNA located 25,000 base pairs downstream of a gene called PTF1A, far outside any coding region. Six different mutations in that single small stretch were the most common cause of isolated pancreatic agenesis they had ever identified.
The PTF1A gene itself was fine. The protein it makes was fine. But the regulatory switch, a tiny enhancer that told the developing embryo when and where to build a pancreas, was broken. One wrong letter was enough.
That finding wasn't an anomaly. It turned out to be the rule.
Today, we know that the vast majority of disease-linked genetic variants don't sit inside genes.[2][3] They sit in the spaces between them, in regions that don't encode any protein, that don't show up in a standard genetic test, that most models can't interpret at all.
This is the problem AlphaGenome was built to solve. But to understand why it's so hard, you first need to understand how genes are actually controlled.
The operating system running on your DNA
Genes don't just switch themselves on. Something has to flip the switch. And those switches aren't the genes themselves, they're specialized DNA sequences scattered across the genome, often sitting far from the gene they control. They're called cis-regulatory elements and you can think of them as the operating system running on top of the genetic hardware.
The main types:
- Promoters sit just upstream of a gene. They're the landing pad, the spot where the molecular machinery that "reads" a gene has to dock before it can start transcribing.
- Enhancers are stranger. They can sit hundreds of thousands of base pairs away from the gene they control, looping through three-dimensional space[4] to touch the promoter and boost expression. An enhancer controlling a gene in your liver cells might be physically closer in space than it is along the linear genome.
- Silencers do the opposite. They dampen expression, or shut it down entirely.
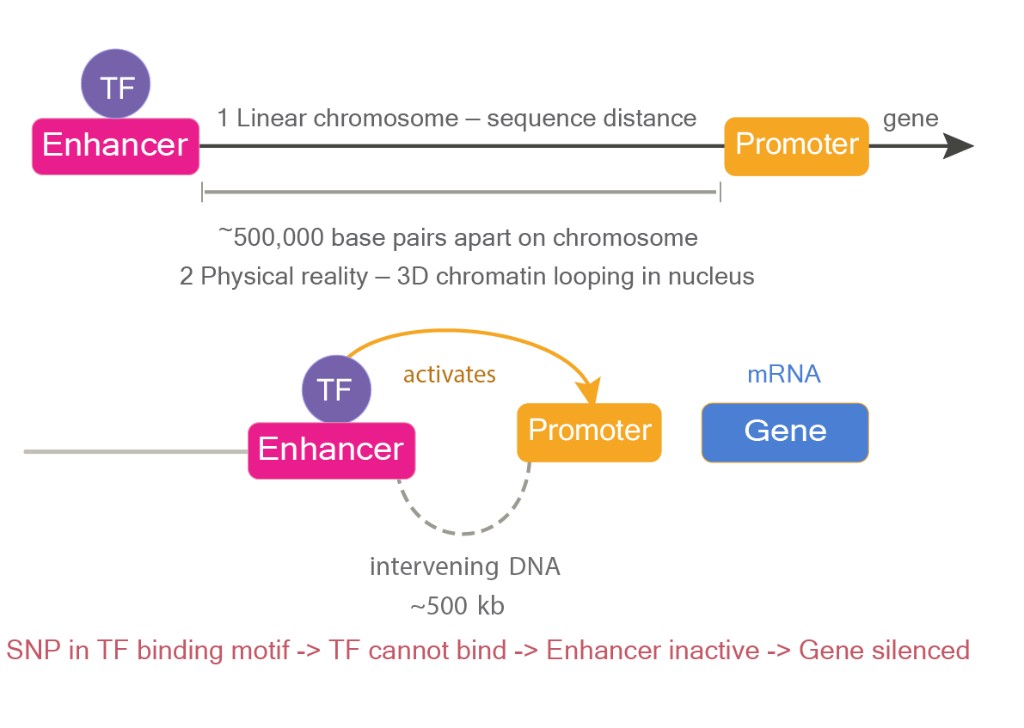
The proteins that actually bind to these regulatory sequences are called transcription factors. Each one recognizes a short, specific stretch of DNA, typically 6 to 20 base pairs long.[5] When a transcription factor finds its motif, it binds. When it binds, it either recruits the transcription machinery or repels it.
Now here's the part that matters for disease.
A single base pair change inside a transcription factor binding site can abolish binding entirely.[2][5] The gene itself is fine. The protein it makes is fine. But the regulatory switch is broken. The gene now gets expressed in the wrong tissue. At the wrong time. At the wrong level.
That's enough to cause disease.
This is why studying the regulatory genome matters as much as studying genes themselves. And it's exactly the problem every genomics model has struggled to crack, because cracking it requires holding two things in your head at once that seem fundamentally incompatible.
The tension that broke every previous model
To model gene regulation computationally, you need two things simultaneously.
Long sequences. An enhancer controlling a gene might sit 500,000 base pairs away. If your model's input window doesn't cover that distance, it can never learn the relationship between them.
Single-nucleotide resolution. A transcription factor binding site is 6 to 12 base pairs long. If you want to know whether a specific variant disrupts binding, you need to see the exact base. You can't average over the sequence.
The problem: longer sequences at finer resolution means exponentially more computation. Every genomics model before AlphaGenome had to pick a side.
Wide window, coarse resolution. Or fine resolution, tiny window.
Neither could do what you actually need, see a 500 kb regulatory landscape and resolve a 6 bp binding motif simultaneously.
AlphaGenome's central contribution is solving that tradeoff. But before we get into how (that's Part 2), it helps to understand the two computational ideas it's built from. Because understanding those ideas is what makes the architecture feel inevitable rather than arbitrary.
Building block 1: CNNs as molecular pattern detectors
Think about what a transcription factor actually does.
It doesn't read the whole genome at once. It diffuses along the DNA, sampling short stretches, lighting up when it finds its specific motif. It's a pattern detector, local, precise, blind to anything outside its small window.
A convolutional neural network filter works the same way.[6]
It slides a small window across the sequence, computing a score at each position. High score means the pattern is there. Low score means it's not. The output, one score per position, is called a feature map. And instead of one filter looking for one pattern, a CNN runs hundreds of them in parallel, each learning a different motif from the data.
CNNs also downsample: 1 Mb of bases → compressed feature map (faster for the next layer). Each position can represent a window of sequence, not only a single base.
Some end up resembling known transcription factor binding sites.[7] Some capture splice sites, the exact base pairs where an exon ends and an intron begins. Some find patterns that biologists haven't formally named yet.
What makes stacked CNN layers powerful is that each layer reads the output of the previous one.
The first layer detects atomic patterns in raw DNA, a TATA box, a splice donor signal, a CTCF motif. The second layer detects combinations: a TATA box plus an initiator element at the right spacing. Later layers can detect higher-order structure, a dense cluster of TF motifs that looks like an active enhancer, or a CTCF site marking a chromatin domain boundary.
By the time the sequence passes through several CNN layers, each position is no longer represented as a simple A, C, G, or T. It carries a rich vector of scores answering questions like: How enhancer-like is the local sequence? Is there a splice site here? A CTCF boundary signal?
CNNs are excellent at this local vocabulary. But they have a hard ceiling.
A CNN filter only sees a short window at a time. It can tell you what's happening at a given position, but it cannot connect an enhancer 400 kb away to the promoter it regulates. For that, you need something different.
Building block 2: Transformers as long-range communicators
A transformer's core mechanism is called self-attention. Here's the intuition.
Every position in the sequence simultaneously generates three things: a question ("what am I looking for?"), a label ("what do I contain?") and a message ("what do I broadcast if someone's interested?").[8] The model then computes a score between every pair of positions. High score means high attention, one position actively incorporating another's information into its own representation.
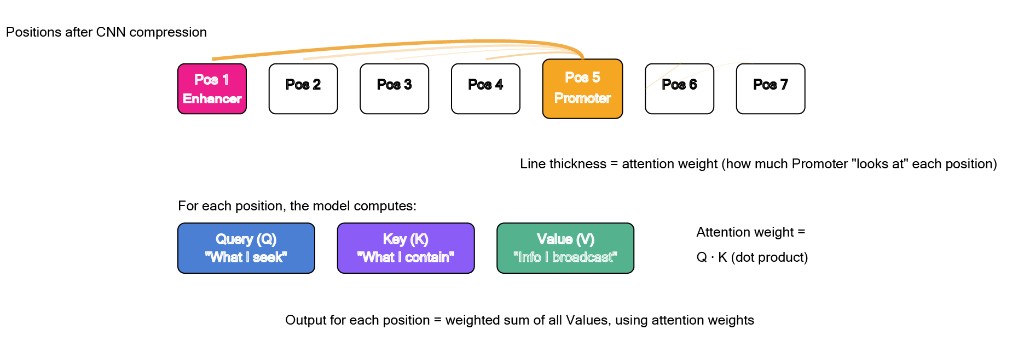
In genomic terms: this is what lets a promoter "attend to" an enhancer 400 kb away and incorporate its regulatory signal. The model learns which positions are functionally related based purely on patterns in the data, no hard-coded rules about which regulatory elements talk to which.
The limitation is cost. Attention scales quadratically with sequence length. Double the sequence, quadruple the computation. At nucleotide resolution across 1 million base pairs, a transformer running directly is simply not feasible.[7][8]
So you can't just bolt a transformer onto the end of a CNN and call it done.
The architectural insight is how you combine them and that's exactly where AlphaGenome does something novel.
What comes next
Two building blocks. One fundamental tension. And a model that, on paper, shouldn't be able to do what it does.
In Part 2: The Architecture, the Model and What It Can Do, we walk through the U-Net architecture that lets AlphaGenome process 1 million base pairs at single-nucleotide resolution[9], why the compress-then-expand structure is the key insight, how skip connections recover fine-grained detail that would otherwise be lost and what the model's 5,930 output tracks[9] actually predict.
References
- [1] Weedon MN et al. "Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis." Nature Genetics, 46, 61–64, 2014. https://doi.org/10.1038/ng.2826
- [2] Bomba L et al. "Decoding Non-coding Variants: Recent Approaches to Studying Their Role in Gene Regulation and Human Diseases." Frontiers in Bioscience, 16(1), 2024. https://doi.org/10.31083/j.fbs1601004
- [3] Sinnott-Armstrong N et al. "The landscape of GWAS validation; systematic review identifying 309 validated non-coding variants across 130 human diseases." BMC Medical Genomics, 15, 2022. https://doi.org/10.1186/s12920-022-01216-w
- [4] Morales J et al. "Understanding the function of regulatory DNA interactions in the interpretation of non-coding GWAS variants." Frontiers in Cell and Developmental Biology, 10, 2022. https://doi.org/10.3389/fcell.2022.957292
- [5] Lambert SA et al. "The Human Transcription Factors." Cell, 172(4), 650–665, 2018. https://doi.org/10.1016/j.cell.2018.01.029
- [6] Kelley DR et al. "Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks." Genome Research, 26(7), 990–999, 2016. https://doi.org/10.1101/gr.200535.115
- [7] Avsec Ž et al. "Effective gene expression prediction from sequence by integrating long-range interactions." Nature Methods, 18, 1196–1203, 2021. https://doi.org/10.1038/s41592-021-01252-x
- [8] Vaswani A et al. "Attention Is All You Need." NeurIPS, 2017. https://arxiv.org/abs/1706.03762
- [9] Avsec Ž et al. "Advancing regulatory variant effect prediction with AlphaGenome." Nature, 2026 (Published 28 January 2026). https://doi.org/10.1038/s41586-025-10014-0