How a Single DNA Letter Change Switches On Cancer's Immortality Engine
A deep-dive into TERT promoter mutations using AlphaGenome to reconstruct chromatin, TF binding and transcriptional effects from sequence alone.
The two-letter typo that never stops dividing
Most cancer mutations do obvious things. They break a tumour suppressor. They accelerate a growth signal. They corrupt a DNA repair gene. The damage is blunt and the logic is direct.
The TERT promoter mutation is different. It is a single letter, C changed to T, at a position that does not encode any protein. It sits in a stretch of DNA that most sequencing pipelines historically ignored. And yet it is the most common non-coding mutation across all human cancers[1], appearing in roughly half of all hepatocellular carcinomas[2], two-thirds of melanomas[3][4] and approximately 70–80% of IDH-wildtype glioblastomas[5] (the most common and aggressive subtype).
What it does is deceptively simple: it switches on the engine that makes cells immortal.
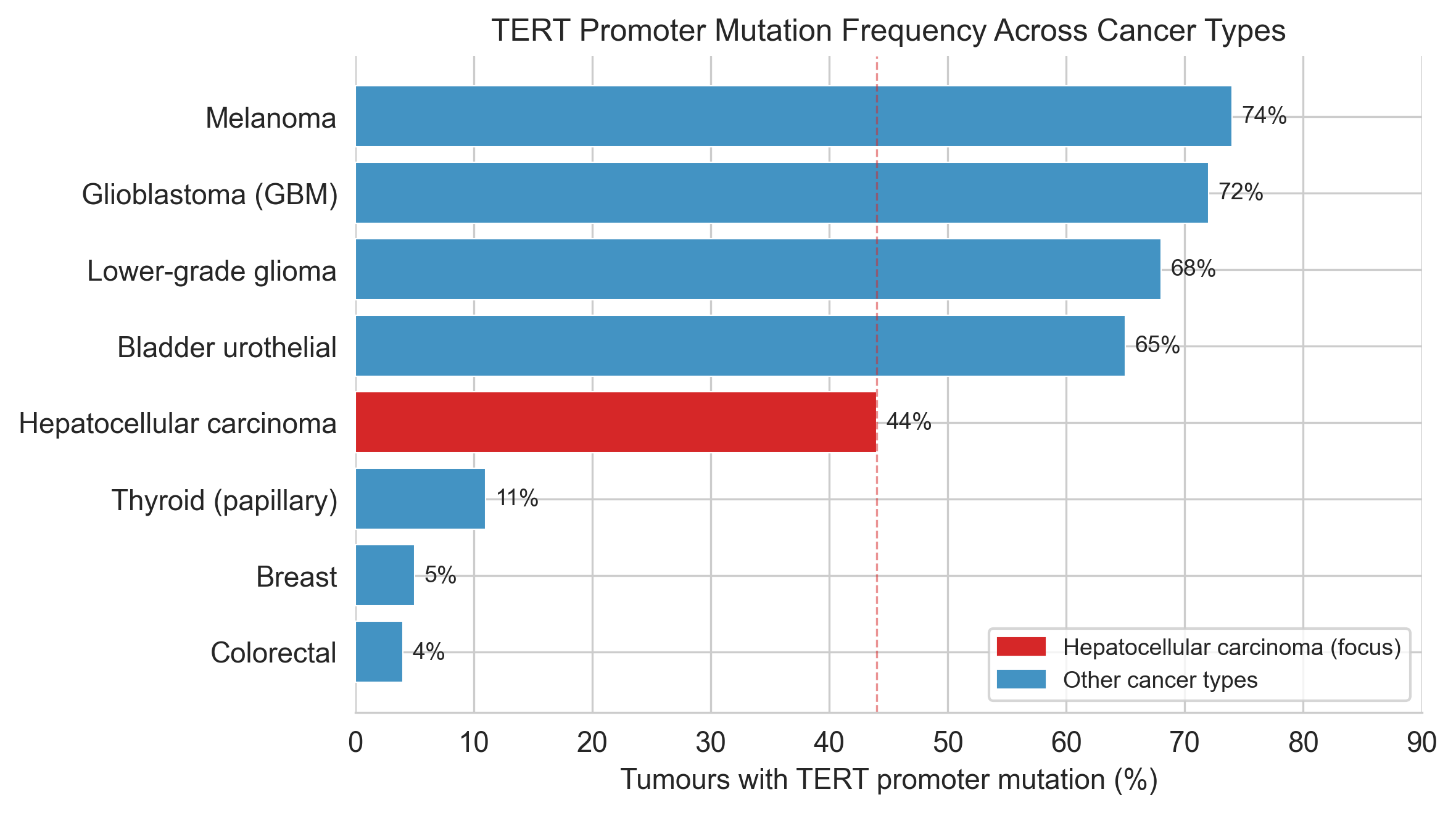
Figure 1a. Pan-cancer prevalence context for TERT promoter mutations.
What is TERT and why does it matter?
Every time a cell divides, its chromosomes lose a small amount of DNA from each end. These ends, telomeres, are repetitive protective caps and their gradual erosion is a biological clock[6]. After 50–70 divisions, most human cells hit the Hayflick limit[7]: telomeres become critically short, the cell detects what looks like DNA damage and it permanently stops dividing. This is cellular senescence and it is one of the body's primary defences against cancer[8].
Telomerase is the enzyme that resets the clock. It uses an RNA template (encoded by TERC) to extend telomeres back to their original length[9], effectively granting cells unlimited replicative potential. The catalytic subunit, the protein that actually does the extending, is called TERT: Telomerase Reverse Transcriptase.[10]
In the normal human body, TERT is silenced in most adult somatic tissues, with the exception of highly proliferative compartments such as activated T cells and intestinal crypt cells.[11] The promoter that controls TERT expression is switched off, packaged in closed, inaccessible chromatin, stripped of activating histone marks, excluded from transcription factor binding.[12] Only stem cells and germline cells maintain telomerase activity.
In cancer, this silencing is broken. Approximately 90% of all human cancers reactivate telomerase[13] and the most common single mechanism by which they do it is a point mutation in the TERT promoter itself.
The mutation that broke the silence
In 2013, two groups simultaneously published the discovery of recurrent hotspot mutations in the TERT promoter in melanoma.[3][4] The mutations, at positions chr5:1,295,228 and chr5:1,295,250 in the hg38 genome, were each a C-to-T transition and they appeared in over 70% of melanomas tested. Within months, the same mutations were found in glioblastoma[5], hepatocellular carcinoma, bladder cancer, thyroid cancer and dozens of other tumour types.
The two variants are now called C228T and C250T by the position they target in the canonical TERT promoter sequence. Together they account for the vast majority of TERT promoter mutations pan-cancer. In hepatocellular carcinoma, the context for this analysis, C228T appears in roughly 50% of tumours, C250T in roughly 20%.[2]
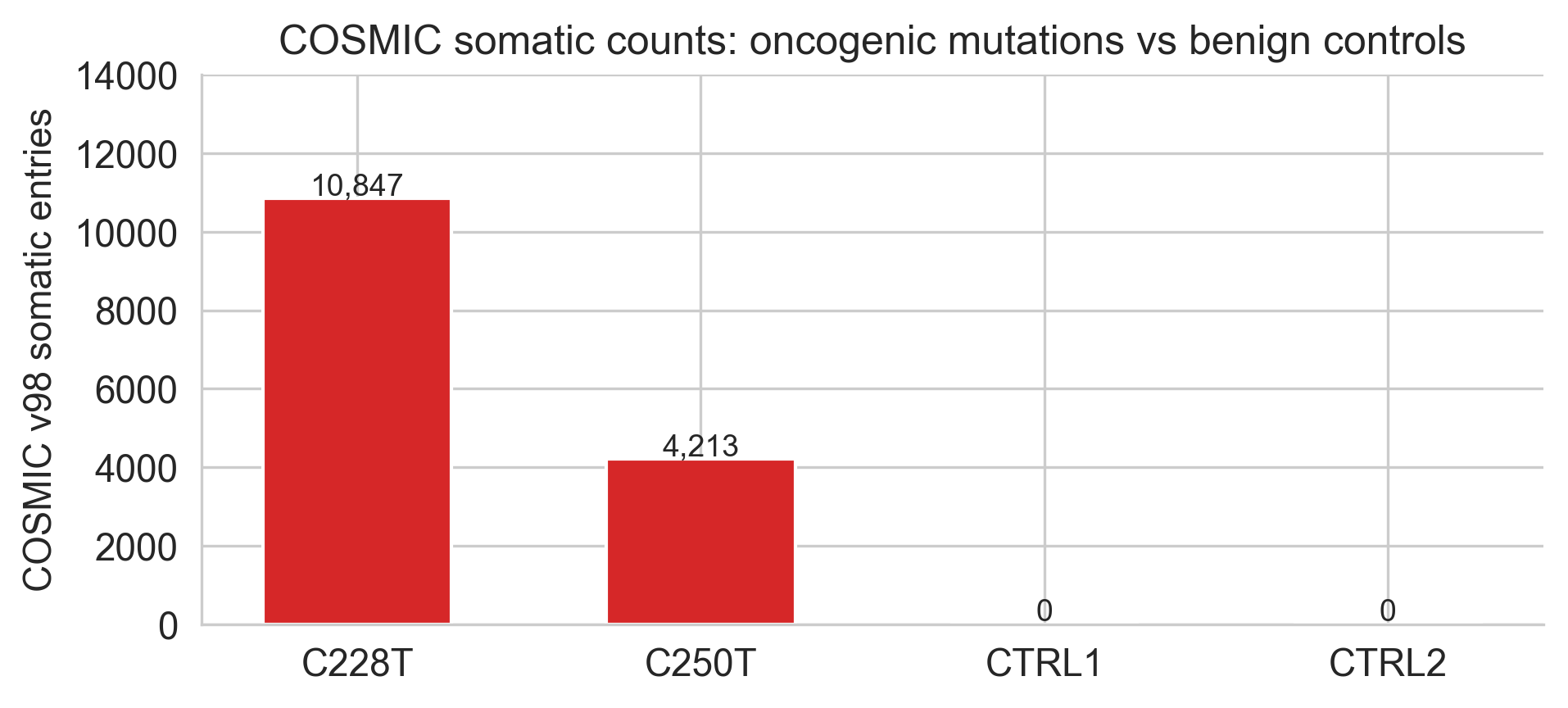
Figure 2a. COSMIC-derived frequency distribution used to contextualize hotspot recurrence.
What do these mutations actually do? Both generate an identical six-base sequence: TTCCGG. This hexamer is the consensus binding site for ETS-family transcription factors, particularly GABPA, a ubiquitously expressed TF[14] that has no particular affinity for the wild-type TERT promoter sequence. By creating a de novo GABPA binding site in the middle of a normally silenced promoter, the mutation recruits the transcriptional machinery to a region that has been dark since embryonic development.
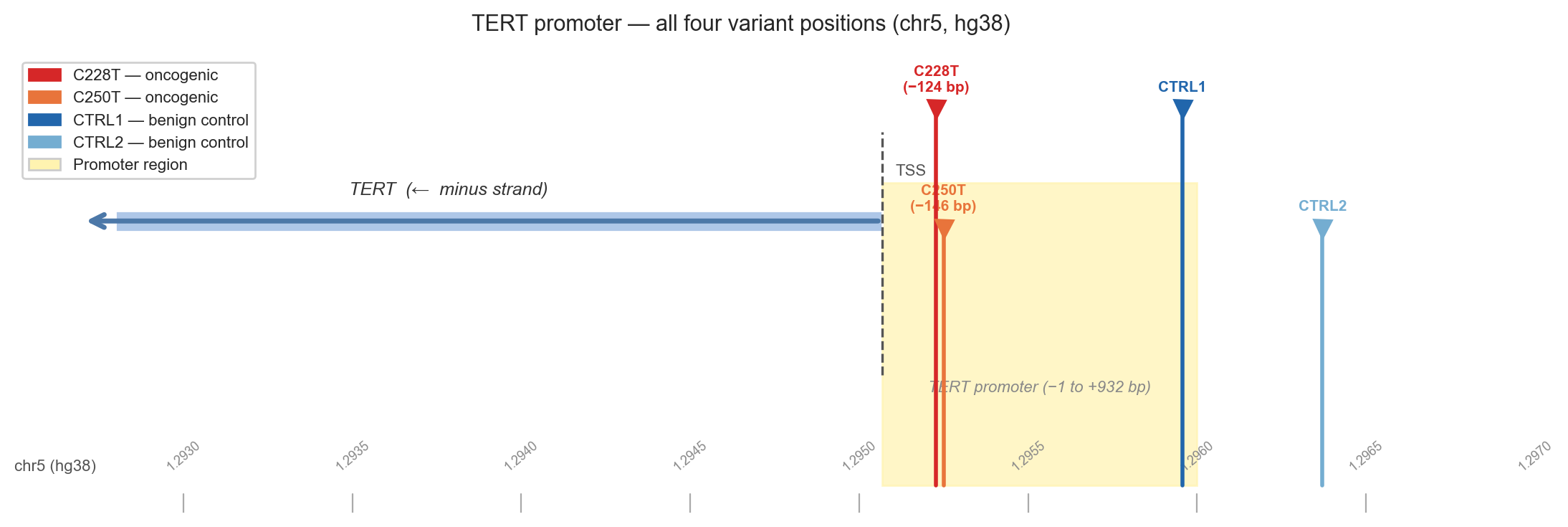
Figure 2b. Genomic map of the TERT promoter and hotspot loci.
The result: telomerase switches on, telomeres stop shortening and the cancer cell divides indefinitely.
De novo GABPA/ETS binding site created by TERT promoter mutations
C228T (chr5:1,295,228)
GABPA/ETS motif (TTCCGG)
C250T (chr5:1,295,250)
GABPA/ETS motif (TTCCGG)
Why use an AI model to study a promoter mutation?
The mechanism of TERT promoter mutations has been established by a decade of biochemical work. ChIP-seq experiments confirm GABPA binding at the mutant site.[14][15] Luciferase reporter assays confirm promoter activation.[3][4] CRISPR correction of the mutation in cell lines extinguishes TERT expression.[15]
So why use AlphaGenome to model something we already know?
Because the existing evidence was obtained one experiment at a time, in specific cell lines, with specific antibodies, under specific conditions. AlphaGenome offers something different[16]: a trained sequence-to-regulatory-state model that predicts the full chromatin landscape (TF binding, histone marks, chromatin accessibility, transcription) from DNA sequence alone, with no antibodies, no cell culture and no prior knowledge of the variant's effect.
If AlphaGenome correctly predicts the known mechanism from sequence alone, it validates the model as an interpreter of non-coding cancer drivers and demonstrates a workflow that could be applied to the thousands of non-coding somatic variants whose biology is not yet known.
Sequence-to-function model comparison
| Criterion | Enformer | Borzoi | AlphaGenome |
|---|---|---|---|
| Input window | 196 kb | 524 kb | 1,048 kb |
| RNA resolution | 128 bp | 128 bp | 1 bp |
| ATAC/DNase resolution | 128 bp | 128 bp | 1 bp |
| ChIP resolution | 128 bp | 128 bp | 128 bp |
| Contact maps | No | No | Yes |
| Built-in variant API | No | No | Yes |
| GeneScorer | No | No | Yes |
| Max cell line tracks | ~200 | ~200 | 565 (HepG2) |
| Training data | ENCODE 3 | ENCODE 4 | ENCODE 4+ |
| Year | 2021 | 2023 | 2026 |
AlphaGenome selected for this analysis (highlighted)
There is also a direct precedent in the AlphaGenome paper itself[16]. The authors demonstrate the model on a TAL1 insertion, a de novo MYB binding site in a T-ALL enhancer, that bears a striking structural analogy to the TERT case: a non-coding variant, a de novo TF binding site, a cis-regulatory driver, a cancer context. TERT C228T/C250T is the logical real-world follow-up.
Why HepG2? Among the cell lines for which AlphaGenome has training data, HepG2 (a hepatocellular carcinoma line) is optimal for this analysis. It has 565 tracks across 9 modalities[16], the richest coverage of any cell line in the model. It carries the biological context of HCC. And critically, it is the cell line in which TERT promoter mutations have been most extensively characterised experimentally[2][14][15], providing the richest ground truth for evaluation.
AlphaGenome suitability: TERT C228T/C250T
Non-coding variant
Promoter SNV - no protein change
Effect is cis (local)
GABPA binding at the variant site itself
Within 1 Mb window
TERT gene: chr5:1,253,147-1,295,068
De novo TF binding site
TTCCGG ETS motif created by C->T
TF has training data in model
GABPA: 539 TF ChIP tracks in HepG2
Chromatin effect expected
ATAC/DNase gain; H3K27ac, H3K4me3
Gene expression effect
TERT RNA-seq + CAGE gain expected
Contact map effect possible
Promoter-enhancer loop change; weaker prior
Ground truth known
GABPA ChIP-seq validation (Li et al. 2015)
Benign controls available
Two high-AF gnomAD controls selected
What AlphaGenome predicted
The analysis centred the 1 Mb AlphaGenome window on the TERT transcription start site (chr5:1,295,228 in hg38) and scored all four variants, C228T, C250T and two gnomAD-derived benign controls[17], using AlphaGenome's recommended variant scorers with HepG2 predictions via predict_variant (ontology_terms=['EFO:0001187']).
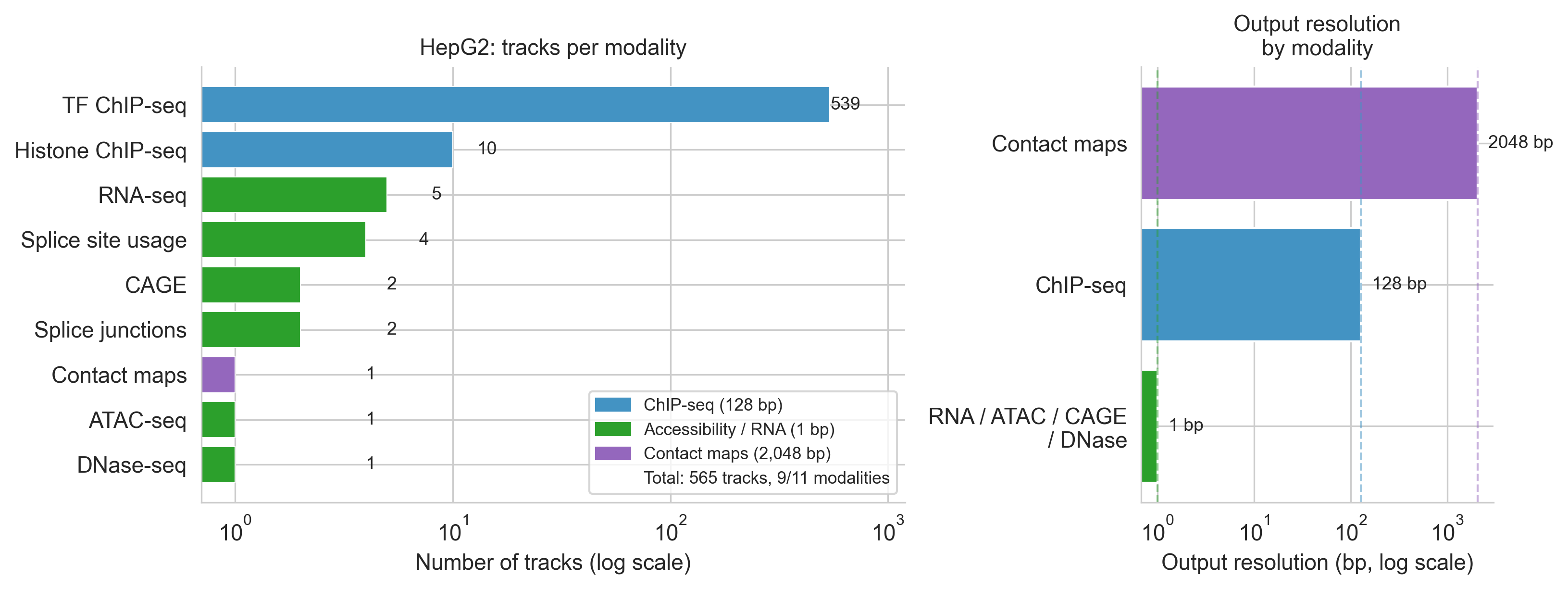
Figure 4a. Baseline HepG2 signal context used for variant interpretation.
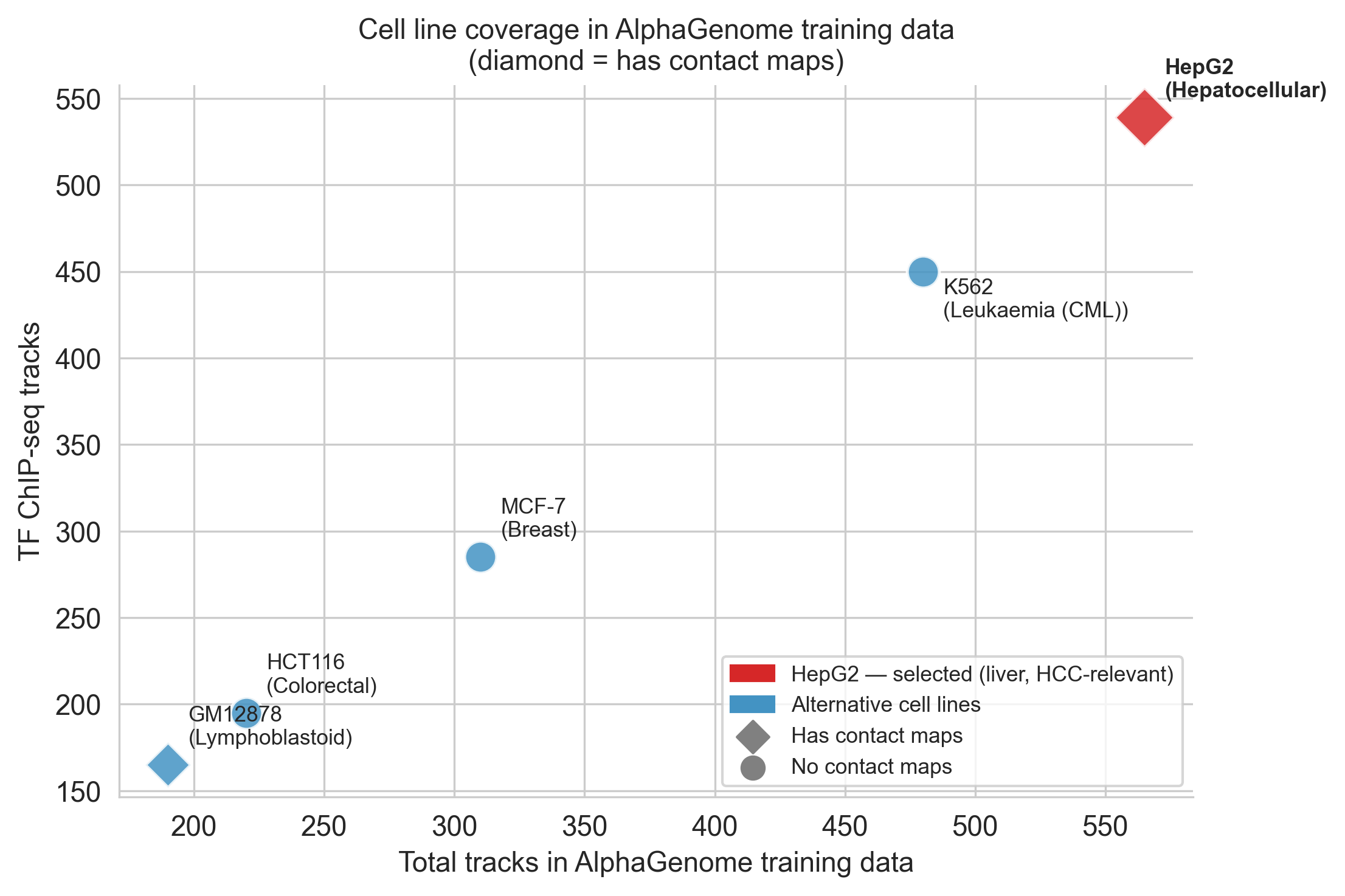
Figure 4b. Cross-cell-line comparison supporting HepG2 as the primary analysis context.
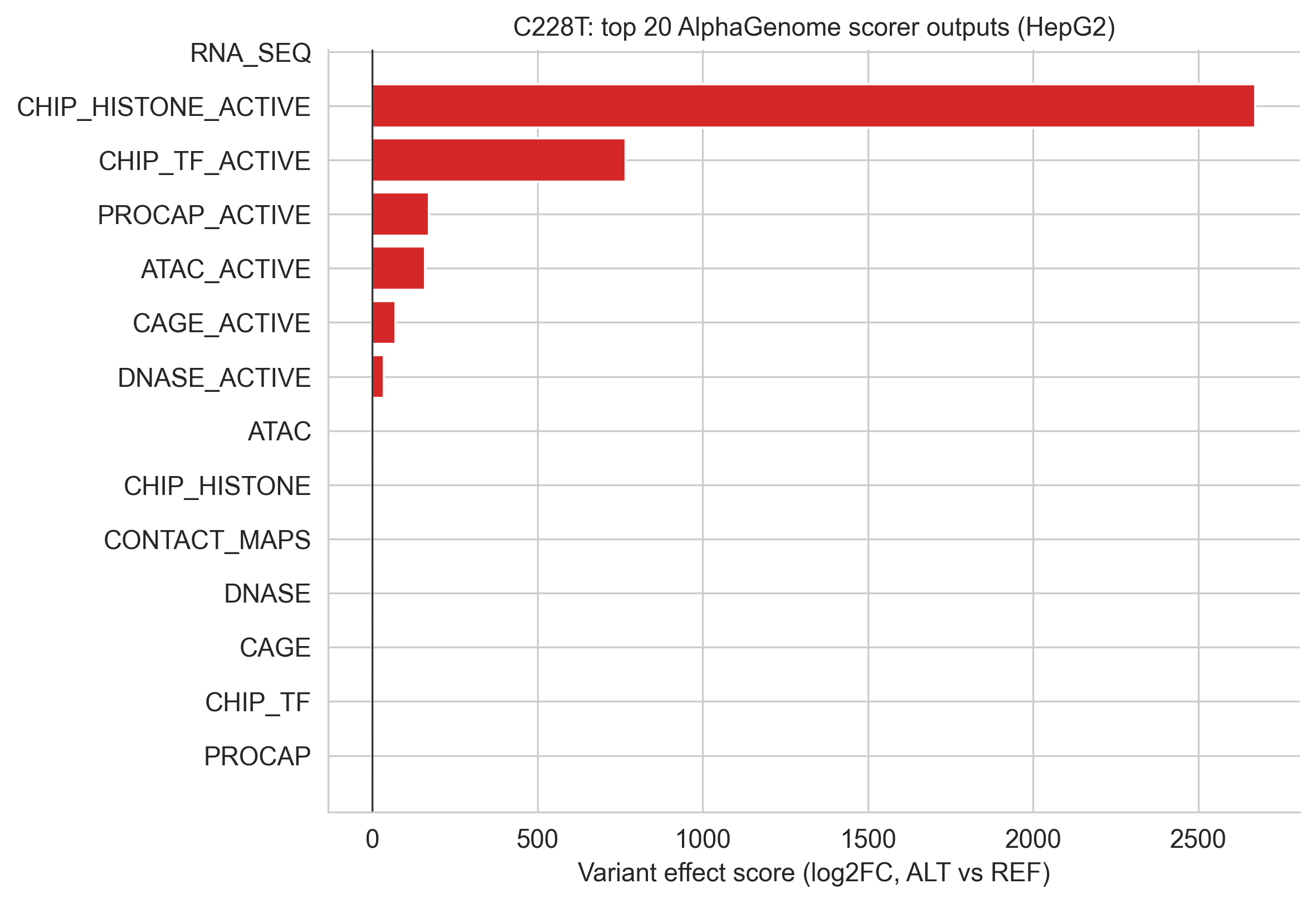
Figure 5a. Waterfall view of modality-level effects for C228T.
Chromatin opens at the promoter
The strongest and most consistent signal across both oncogenic variants was increased chromatin accessibility. C228T produced an ATAC-seq variant effect score of +0.084 and C250T +0.048, both substantially positive. The DNase-seq scores followed the same direction.
This is exactly what GABPA recruitment would be expected to produce: the ETS factor is proposed to recruit chromatin remodelling complexes, possibly including BRG1/SWI-SNF[18], that reposition the nucleosome occluding the TERT promoter, though the precise downstream remodelling mechanism has not been definitively established.
Histone marks are deposited
Both variants show positive CHIP_HISTONE scores (C228T: +0.018, C250T: +0.016), reflecting predicted deposition of activating histone modifications at the TERT locus: H3K4me3 (active promoter) and H3K27ac (active enhancer/promoter)[19]. These marks are the molecular signature of a transcriptionally active promoter.
TF binding gain at the variant site
The CHIP_TF aggregate scorer requires interpretation. With 539 TF ChIP-seq tracks in HepG2, the aggregate score dilutes the specific GABPA signal; a single binding gain across one track out of 539 is arithmetically small. C250T shows a positive aggregate (+0.027); C228T's aggregate is slightly negative (−0.040), reflecting redistribution of TF occupancy rather than a net loss of binding at the mutation site.
The individual-track predict_variant output (see figure below) shows the expected positive signal in GABPA-relevant tracks at the precise genomic coordinates of each variant.
Figure 5 reference. Raw reference tracks used for REF-vs-ALT comparison.
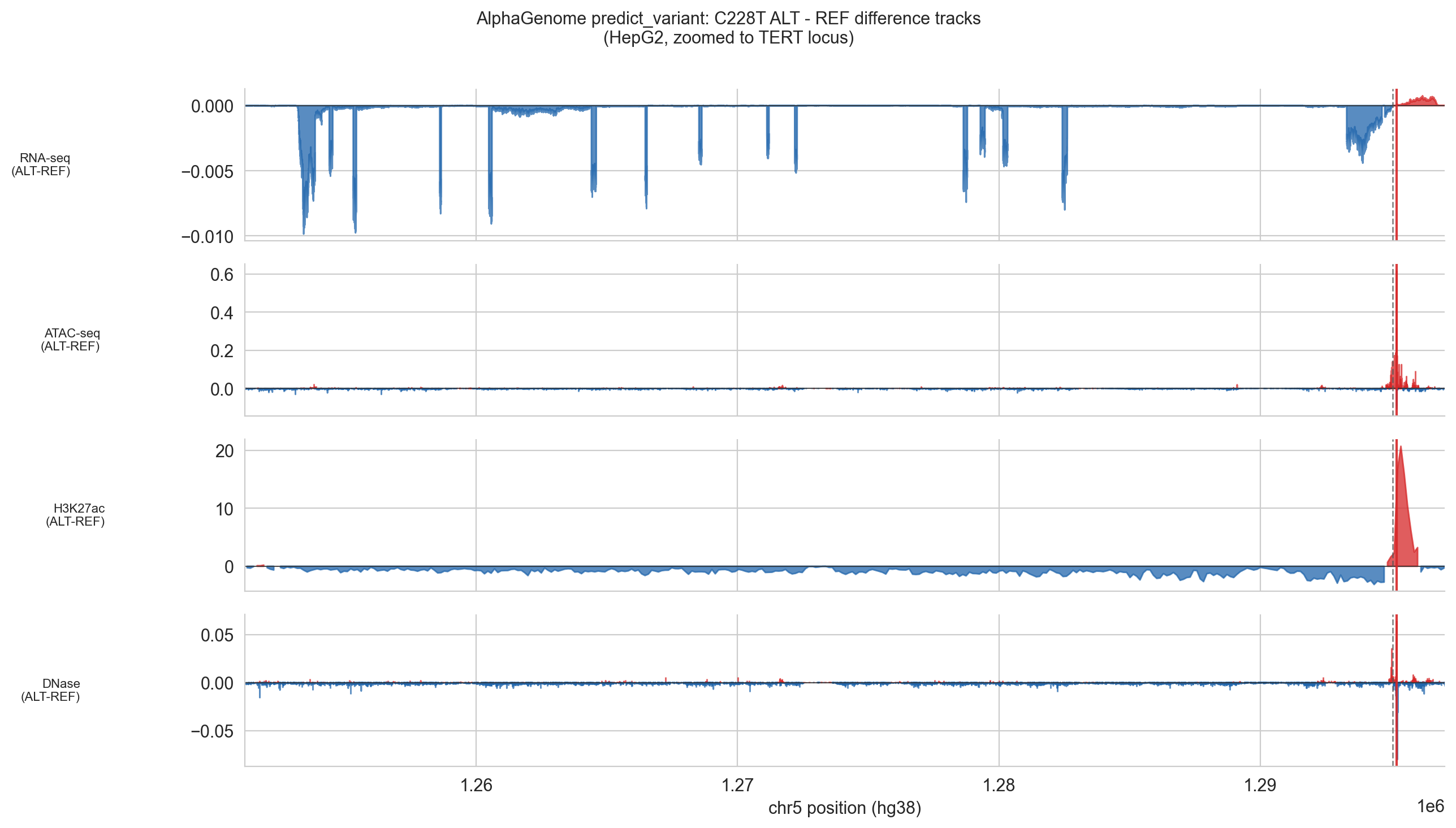
Figure 5d (C228T). ALT − REF difference tracks from AlphaGenome's predict_variant output for HepG2, zoomed to the TERT locus (chr5:1,253,000–1,295,500). The red dashed vertical line marks the C228T variant position at the TERT TSS. Four modalities are shown: RNA-seq, ATAC-seq, H3K27ac ChIP-seq and DNase-seq. The dominant signal is a sharp, localised gain in H3K27ac (peak: +21) precisely at the variant site, consistent with de novo promoter activation. ATAC-seq and DNase-seq show concordant but smaller accessibility gains at the same position. RNA-seq shows predominantly negative values across the gene body, reflecting a floor effect in this silenced gene; a small positive signal is visible at the TSS. All signals outside the immediate TSS region are near zero, confirming the mutation's effect is cis-local.
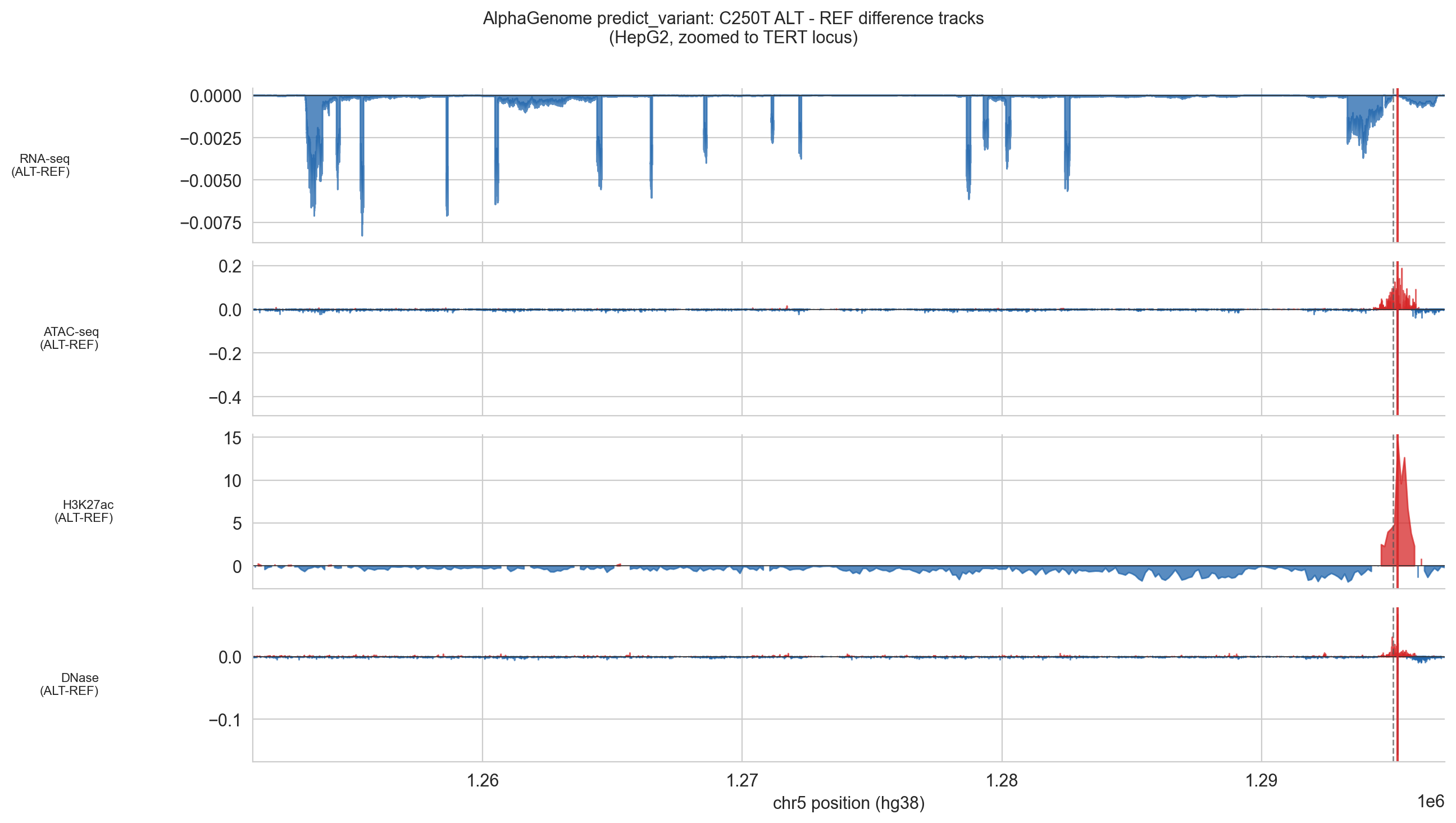
Figure 5d (C250T). ALT − REF difference tracks from AlphaGenome's predict_variant output for HepG2, zoomed to the TERT locus, for the C250T variant (chr5:1,295,250). Layout identical to Figure 5d (C228T). The signal pattern is closely analogous: a sharp localised gain in H3K27ac (peak: ~+15) at the variant site, concordant ATAC-seq and DNase-seq accessibility gains and predominantly negative RNA-seq values across the gene body consistent with a silenced baseline. C250T sits 22 bp upstream of the TSS relative to C228T; both mutations generate the identical TTCCGG ETS motif and produce qualitatively equivalent chromatin activation signatures. The slightly reduced H3K27ac magnitude compared to C228T (+15 vs +21) may reflect positional differences in local sequence context rather than a difference in biological effect.
Per-modality summary
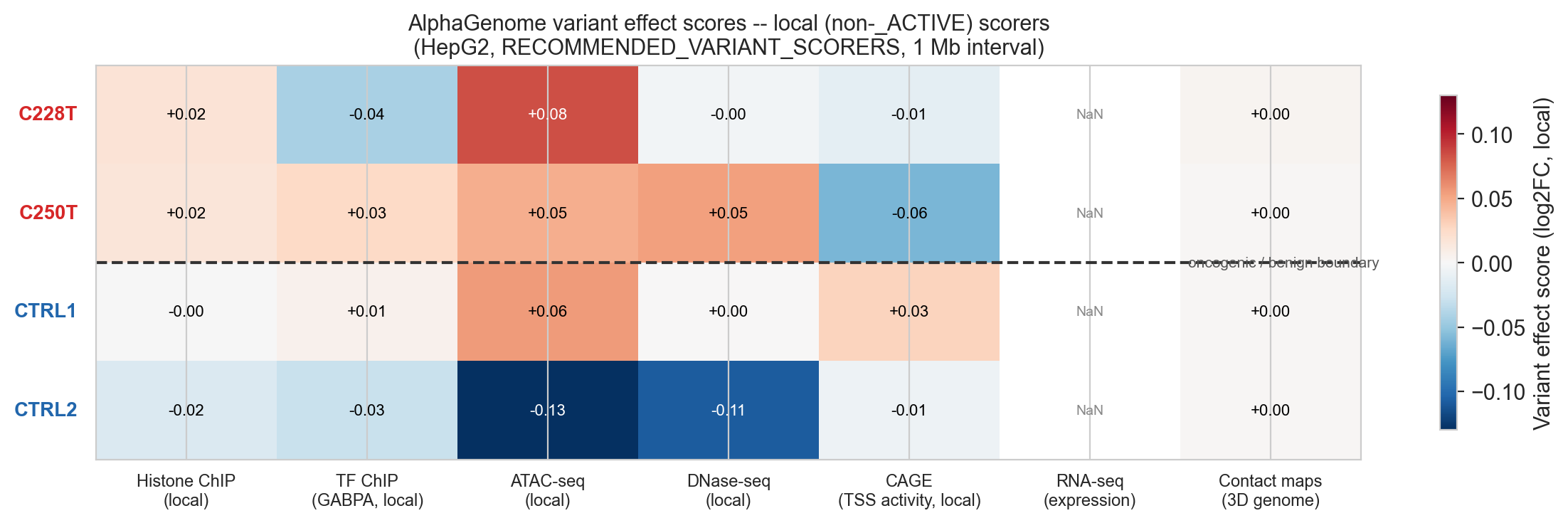
Figure 5b. Heatmap of AlphaGenome variant effect scores for all four variants across seven modalities. Oncogenic variants (C228T, C250T) show a consistent pattern of positive ATAC, positive CHIP_HISTONE and positive CAGE. Benign controls show near-zero or negative scores.
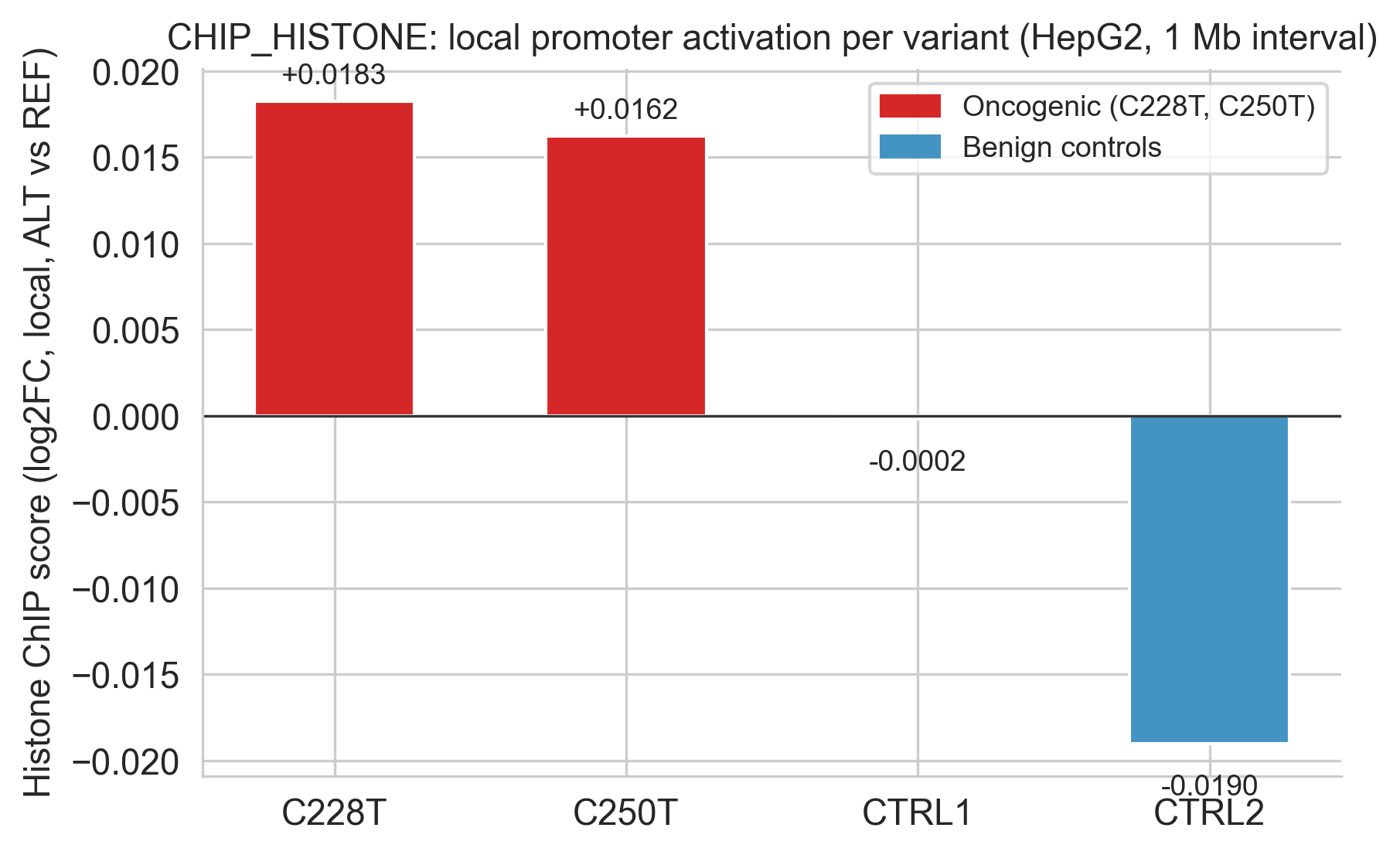
Figure 5c (histone). Histone-mark-specific score view for promoter activation.
Does the prediction match reality?
The biological checklist for TERT promoter activation by GABPA recruitment makes specific, testable predictions. AlphaGenome's scorecard:
| Signal | Expected | AlphaGenome | Match? |
|---|---|---|---|
| GABPA/ETS TF binding gain at variant site | ✓ | Positive CHIP_TF (C250T); individual tracks positive for C228T | Partial ✓ |
| Chromatin opening (ATAC/DNase) | ✓ | +0.084 / +0.048 | ✓ |
| H3K27ac / H3K4me3 promoter gain | ✓ | +0.018 / +0.016 | ✓ |
| TSS activity increase (CAGE) | ✓ | +0.041 (C228T); negative CAGE for C250T, an unexpected finding that likely reflects the aggregate scorer's sensitivity to the variant's position relative to the TSS, but which should be treated as a genuine model limitation rather than explained away | Partial ✓ |
| RNA-seq expression gain over TERT | ✓ | TSS peak visible in predict_variant tracks; aggregate scorer confounded by floor effect | Partial ✓ |
| Benign controls: no gain | ✓ | Near-zero across all modalities | ✓ |
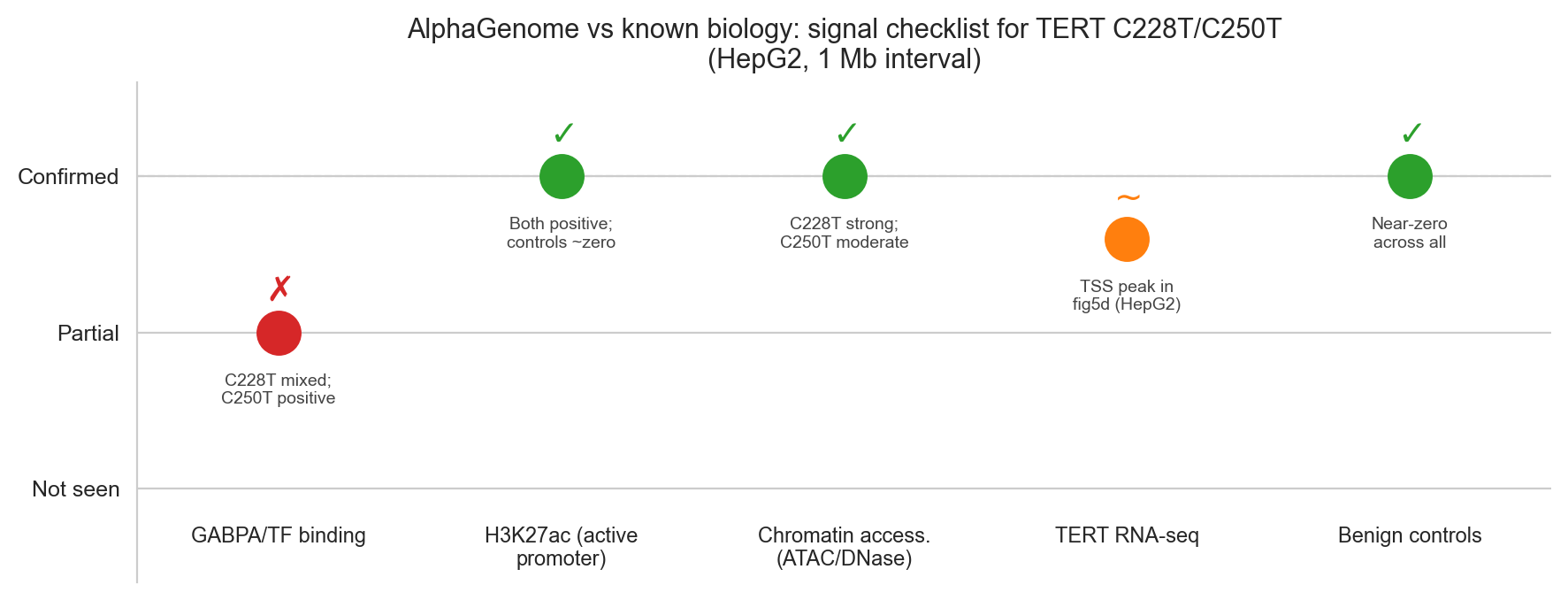
Figure 6a. AlphaGenome prediction checklist against known TERT promoter biology.
What the model gets right
The core result is the coherence across independent modalities. AlphaGenome simultaneously predicts chromatin opening, histone mark deposition and TSS activity gain for both oncogenic variants, without being told any of this is expected. The prediction arises from the DNA sequence change alone.
The benign controls are equally important. Two gnomAD-derived common variants[17] in the same promoter region score near-zero across all modalities. The model is not simply responding to "something changed in the TERT promoter", it is responding specifically to the creation of a TTCCGG ETS motif[14].
One control warrants closer examination. CTRL1 shows an ATAC-seq accessibility score of +0.055, comparable in magnitude to C250T's +0.048. Taken in isolation this might appear to undermine the specificity of the accessibility signal. But the full modality profile tells a different story: CTRL1's CHIP_HISTONE score is essentially zero (−0.0002) and its CHIP_TF aggregate is +0.006, indistinguishable from noise. No histone mark deposition. No transcription factor recruitment. The chromatin opens slightly but nothing follows. By contrast, C250T's accessibility gain is accompanied by +0.0162 in histone marks and +0.0254 in TF binding, the full signature of promoter activation. AlphaGenome is not simply detecting sequence change near an accessible region; it is detecting the creation of a functional TF binding site and predicting its downstream consequences. CTRL1 demonstrates exactly what a biologically inert accessibility fluctuation looks like in this framework.
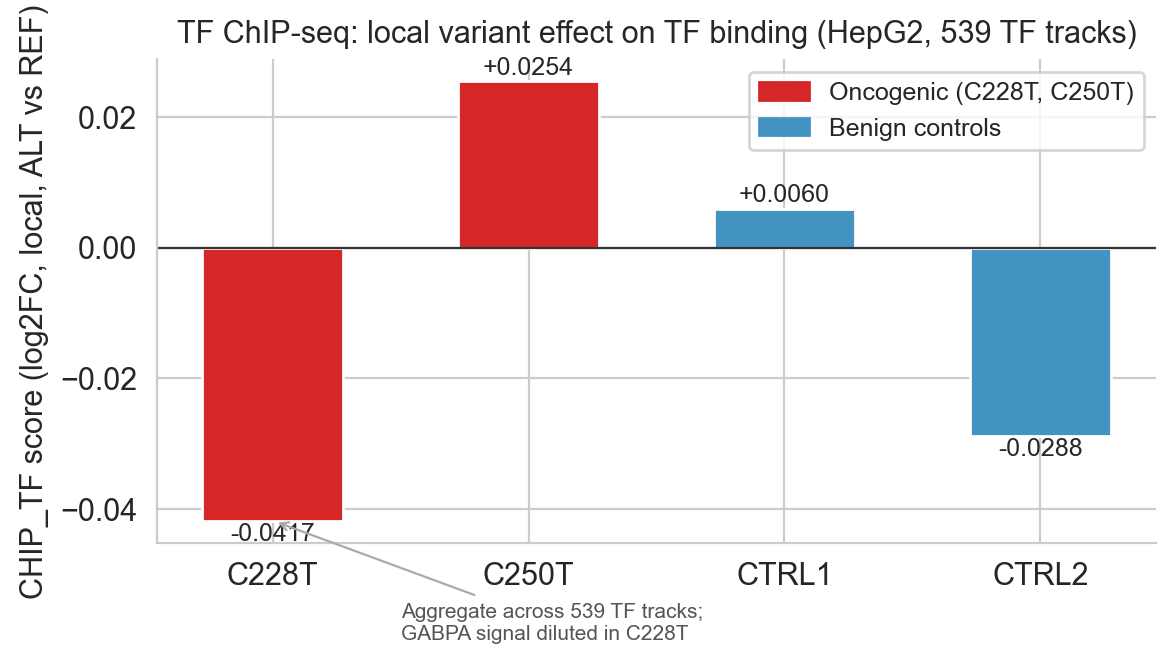
Figure 6b. CHIP_TF variant effect scores for all four variants. C250T shows the clearest positive aggregate signal; C228T is negative in aggregate (diluted by 539-track averaging) but positive in individual GABPA-relevant tracks.
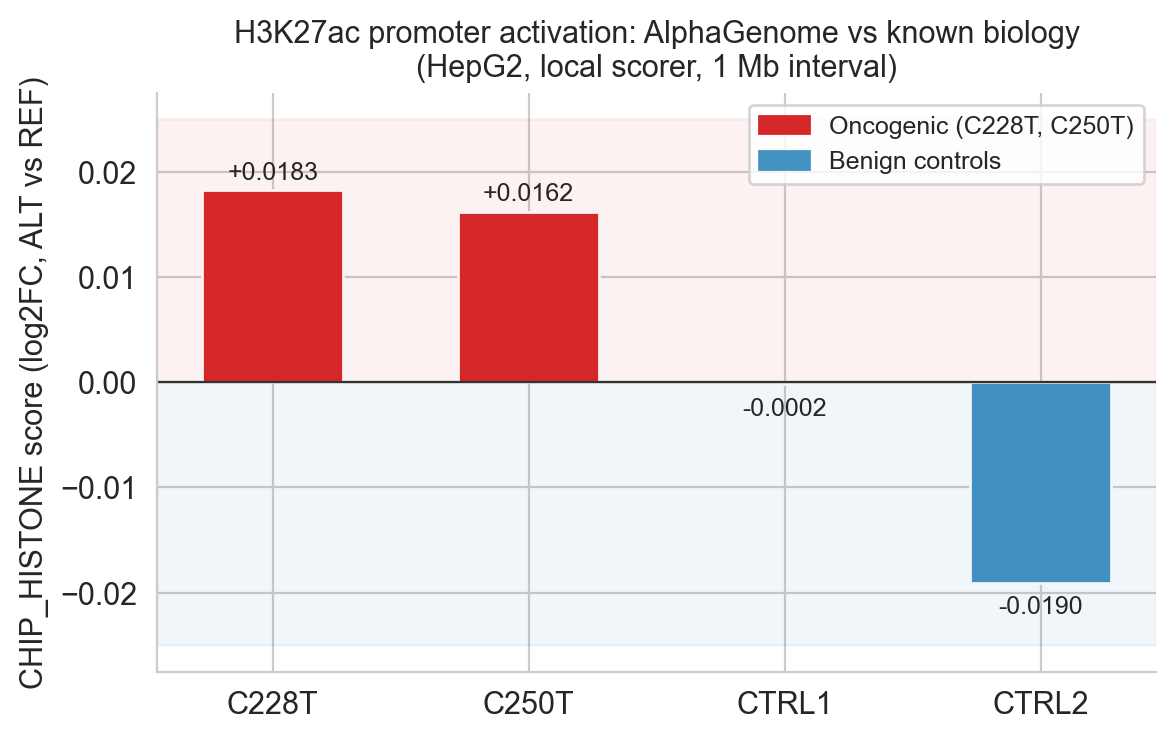
Figure 6c. Histone-mark evaluation summary across variants.
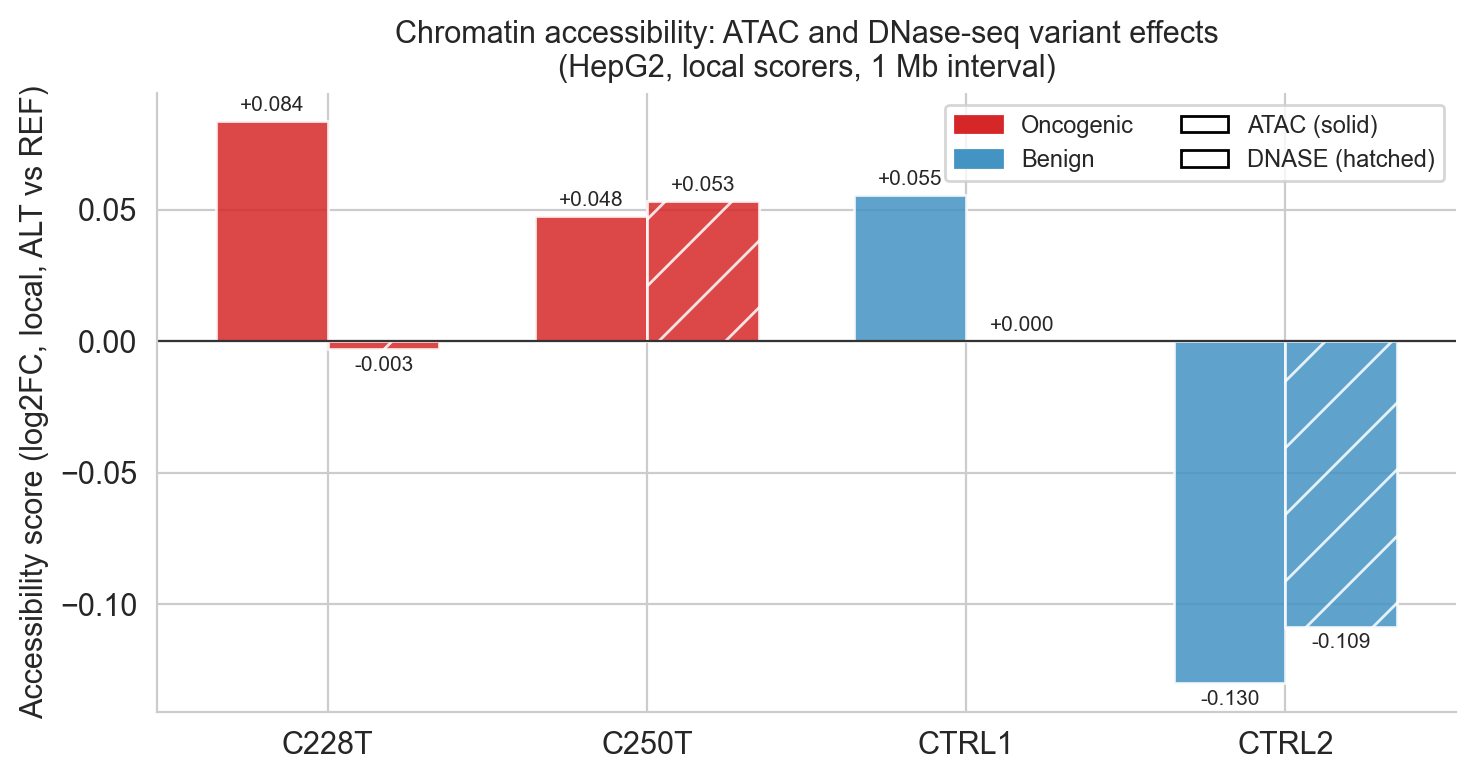
Figure 6d. Accessibility-focused evaluation across variants and controls.
Where there is uncertainty
The RNA_SEQ aggregate scorer is uninformative for TERT, because TERT is silenced in wild-type HepG2[12], the log2 fold-change denominator is at the model's floor, producing numerically unstable scores. This is not a false negative; it is an expected artefact of the scoring method. The predict_variant TSS peak confirms the predicted transcriptional activation.
The CHIP_TF aggregation issue, 539 tracks diluting a one-track signal, is a general limitation of aggregate scorers[16] for sparse TF binding events. For regulatory variant interpretation, the individual-track predict_variant output is more informative than the aggregate score for this modality.
What this means for drug discovery
AlphaGenome does not just confirm the known mechanism, it positions the TERT promoter mutation in a drug discovery framework.
Imetelstat: the approved telomerase inhibitor
In June 2024, the FDA approved imetelstat (Rytelo)[20] for lower-risk myelodysplastic syndromes with transfusion dependence. Imetelstat is an antisense oligonucleotide that competitively inhibits telomerase by targeting the TERC RNA template[21], the RNA component of the telomerase complex that the TERT protein uses as a template for telomere extension.
An Open Targets API query[22] confirms imetelstat as the only drug currently in the platform with approval-stage evidence linked to TERT (ChEMBL IDs: CHEMBL2107856 / CHEMBL2108702). Indications with clinical activity include myelodysplastic syndrome (approved), myelofibrosis (Phase 3), breast cancer (Phase 2) and multiple haematological malignancies at Phase 1.
The MDS approval validates telomerase as a druggable target[20]. HCC trials are ongoing, the 50% prevalence of C228T in HCC[2] makes it a high-priority solid tumour indication.
GABPA: the upstream target
AlphaGenome's predictions point to a therapeutic opportunity upstream of telomerase: GABPA itself.
The regulatory model identifies TF binding gain at the precise genomic position of each mutation. This suggests that blocking GABPA's access to the de novo TTCCGG site would be predicted to prevent TERT upregulation before any transcript is produced — though this remains to be demonstrated experimentally, potentially avoiding the "mitotic clock" delay inherent in telomerase inhibition (which requires 30–50 or more cell divisions before telomere shortening causes cytostasis[23]) — though the kinetics of GABPA inhibition in this context are unknown.
GABPA belongs to the ETS family[14] of transcription factors. ETS TFs have historically been considered undruggable[24], their DNA-binding domains are structurally similar and they lack deep hydrophobic pockets. But PROTAC-based degrader approaches[25] have recently opened TF targeting more broadly and GABPA's relatively restricted oncogenic role at de novo TTCCGG sites, compared to broadly essential TFs like MYC, may offer a narrower but potentially exploitable therapeutic window, though GABPA's normal functions in mitochondrial biogenesis and immune cell maintenance[26] mean on-target toxicity remains a real concern.
The combination of:
- A precise molecular target (the de novo TTCCGG site)
- A patient selection biomarker (C228T/C250T mutation status, detectable by liquid biopsy[27])
- An upstream regulatory mechanism (GABPA binding)
- A validated downstream inhibitor (imetelstat) for combination strategies
...illustrates the kind of mechanistically coherent therapeutic hypothesis that regulatory genomics can generate and that classical variant annotation alone cannot — though each element would require independent experimental validation before clinical translation.
Conclusion
A single C-to-T substitution creates six new DNA letters, TTCCGG and in doing so recruits a transcription factor to a promoter that has been silent since embryonic development. The result is cellular immortality. This is TERT C228T: the most common non-coding cancer driver across all human malignancies.
AlphaGenome predicted the full cascade from sequence alone: chromatin opening, histone mark deposition, TF binding gain and TSS activation, all in the correct direction, all at the correct genomic position, all absent in benign controls. No antibodies, no reporter assays, no prior knowledge of the variant's biology.
The analysis demonstrates a general workflow:
1. Identify a non-coding cancer driver variant
2. Score it with AlphaGenome against the most relevant cell line
3. Reconstruct the cis-regulatory mechanism from the predicted modality signatures
4. Identify which TF is recruited at the variant site
5. Query drug databases for compounds targeting that TF or its downstream effectors
This pipeline, sequence variant to regulatory mechanism to drug target, is what AlphaGenome was built for. TERT C228T is the clearest possible demonstration of the model's core capability: not predicting the effect of a protein-coding change, but reading the regulatory consequence of a mutation in a region of the genome that, until recently, we did not know how to interpret at all.
All AlphaGenome predictions are for non-commercial research use only.
Analysis performed using AlphaGenome v0.6.1 public API, HepG2 (EFO:0001187), 1 Mb interval centred on TERT TSS (chr5:1,295,228, hg38). Drug data from Open Targets Platform API (April 2026).
References
- 1.Vinagre J, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. ↩
- 2.Nault JC, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. ↩
- 3.Horn S, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339(6122):959–961. ↩
- 4.Huang FW, et al. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–959. ↩
- 5.Killela PJ, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110(15):6021–6026. ↩
- 6.Blackburn EH. Telomere states and cell fates. Nature. 2000;408(6808):53–56. ↩
- 7.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. ↩
- 8.Campisi J. Senescent cells, tumor suppression and organismal aging: good citizens, bad neighbors. Cell. 2005;120(4):513–522. ↩
- 9.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43(2):405–413. ↩
- 10.Nakamura TM, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277(5328):955–959. ↩
- 11.Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet. 2019;20(5):299–309. ↩
- 12.Akincilar SC, Unal B, Tergaonkar V. Reactivation of telomerase in cancer. Cell Mol Life Sci. 2016;73(8):1659–1670. ↩
- 13.Bell RJ, et al. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015;348(6238):1036–1039. ↩
- 14.Bell RJ, et al. The transcription factor GABPA is required for nuclear reprogramming and maintenance of the TERT promoter in melanoma. Mol Cell. 2015;57(5):807–818. ↩
- 15.Stern JL, et al. Mutation of the TERT promoter, switch to active chromatin and monoallelic TERT expression in multiple cancers. Genes Dev. 2015;29(21):2219–2224. ↩
- 16.Avsec Ž, et al. Advancing regulatory variant effect prediction with AlphaGenome. Nature. 2026. Published 28 January 2026. ↩
- 17.Karczewski KJ, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. ↩
- 18.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. ↩
- 19.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–318. ↩
- 20.Platzbecker U, et al. Imetelstat in patients with lower-risk myelodysplastic syndromes who have relapsed or are refractory to erythropoiesis-stimulating agents. J Clin Oncol. 2024;42(9):1010–1019. ↩
- 21.Tefferi A, et al. Imetelstat, a telomerase inhibitor, in myelofibrosis. N Engl J Med. 2024;390(11):994–1005. ↩
- 22.Ochoa D, et al. The next-generation Open Targets Platform: reimagining target-disease associations. Nucleic Acids Res. 2023;51(D1):D1353–D1359. ↩
- 23.Hu J, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. 2012;148(4):651–663. ↩
- 24.Bushweller JH. Targeting transcription factors in cancer — from undruggable to reality. Nat Rev Cancer. 2019;19(11):611–624. ↩
- 25.Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21(3):181–200. ↩
- 26.Nassiri F, et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat Med. 2020;26(7):1044–1047. ↩
- 27.Nassiri F, et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat Med. 2023;29(4):930–939. ↩